Contents
Cation-p Interactions
Snapshots
Salt Bridges
|
Background.
Gallivan & Dougherty (1999)
reported results from a quantitative survey of
cation-p
(cation-pi) interactions in
high-resolution structures in the Protein Data Bank. Using an energy-based
criterion for identifying significant sidechain interactions, they
studied 593 sequence
dissimilar proteins taken from the
"PDB Select" list of Hobohm and Sander.
They found an average of one such interaction per 77 residues, with no
significant effect of chain length, or multiple-chain vs. single chain
structures.
Arg was more
likely than Lys to participate in a cation-pi interaction, and
the liklihood of aromatic sidechain participation
was Trp > Tyr > Phe. Over one quarter of all Trp's were involved in
cation-pi interactions, with the
cation typically positioned over the 6-atom ring of Trp.
Because of the frequencies of amino acids in the database,
Arg participates in nearly twice as many cation-pi interactions as does Lys,
and the numbers of cation-pi interactions involving Trp, Tyr and Phe are
roughly similar.
Their study did not include His because, depending on its protonation state,
it could participate either as a cation or as a pi-system. Lys and Arg
were assumed always to be protonated and hence cationic.
Gallivan and Dougherty conclude "When a cationic sidechain is near
an aromatic sidechain, the geometry is biased toward one that would experience
a favorable cation-pi interaction", and "cation-pi interactions should be considered
alongside the more conventional hydrogen bonds, salt bridges, and hydrophobic
effects in any analysis of protein structure". They provide a
Gallery
of energetically significant cation-pi interactions
and a
server that lists text results from their program
CaPTURE.
Zacharias and Dougherty (2002) reviewed cation-pi
interactions in the binding of ligands to proteins. Cation-pi interactions
are usually energetically
important when the ligand has either positive charge or an aromatic ring,
and are involved in control of ion channels, G-protein-coupled receptors,
transporters, and enzymatic catalysis.
An example is 1L8B, a portion of a eukaryotic
translation initiation factor that recognizes N7-methylated guanosine.
The ligand's heterocyclic base (cationic) is sandwiched between Trp56
and Trp102. The easiest way to see this in PE is in QuickViews: SELECT Ligand, DISPLAY
Contacts. (Alternatively, you can designate the cationic ligand nitrogen in
the Cation-Pi form in
Advanced Explorer. Note that QuickViews DISPLAY Cation-Pi
does not display cation-pi interactions involving ligands.)
An introductory
gallery of examples of cation-pi interactions and other
noncovalent interactions
is provided by Ricky Cox.
Contents
Close
Pros & Cons of Protein Explorer's Cation-Pi Display
As detailed below, Protein Explorer's present
mechanism to display cation-pi interactions tends to show
more cation-pi pairs than the number deemed energetically
significant by Gallivan & Dougherty's
CaPTURE (up to
twice as many pairs). It also sometimes misses one or more pairs
deemed energetically significant by CaPTURE. Therefore,
Protein Explorer's cation-pi display is useful for a first look, but important
cases should then be compared to the output of CaPTURE. (It is planned
eventually to enable CaPTURE to display its output directly in Protein Explorer.)
Protein Explorer allows you to include ligands in the cation-pi display,
a capability that is not implemented in the CaPTURE web server.
In order to include ligand cations or aromatic rings, you must specify
the appropriate atoms in the cation-pi form slots. Several examples
of how to do this are explained
in the tutorials below.
Contents
Close
Interesting Examples and Gallery
Examples given by Gallavan and Dougherty include:
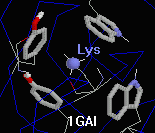
- 1gai: a 472-amino acid chain (glucoamylase) with a
spectacular cluster of four aromatic rings
(two Trp's, two Tyr's) around a single Lys108.
- 1bfg: a 126-amino acid chain (fibroblast growth factor) with
an unusually high incidence of cation-pi interactions. Gallivan and
Dougherty report 5 energetically significant interactions; Protein
Explorer shows 9 rings and 8 cations; one ring interacts with two
cations, making the total interactions shown equal to 10.
- 2wea: the longest single chain (323 residues) with no
energetically significant cation-pi interactions. Despite its
tendency to overestimate, Protein Explorer satisfyingly displays
none.
Interesting examples noted using Protein Explorer:
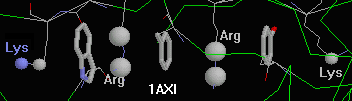
- 1axi (human growth hormone): Chain
B contains an unusual string of three aromatic sidechains separated by, and
capped at the ends with, 4 cationic sidechains. (Using "c" for
cation and "p" for pi, the chain is "cpcpcpc".) Only the one of these six
interactions is deemed energetically insignificant by CaPTURE (Lys at one end).
- 1bl8 (bacterial potassium channel): There is
a single interchain cation-pi pair for each contact between
chains of this homotetramer, but no intrachain cation-pi interactions.
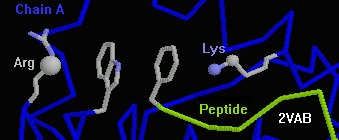
- 2vab (peptide bound to class I histocompatibility protein): The N-terminal
Phe of the nonapeptide stacks with Trp 167 of the protein. On either side
of the stacked rings are cations (Arg 170, Lys 66), forming the unusual
cppc chain RWFK.
- 1rog (peptide bound to class I histocompatibility protein): Three
(of the four) cations in the 9-residue peptide interact with
aromatic sidechains in the protein groove. This is a theoretical
model.
- 1dlh (peptide bound to class II histocompatibility protein):
There are no cation-pi interactions for the 13-residue peptide, despite
its containing three lysines and one tyrosine. A number of nearby
sidechains that potentially could interact appear to be blocked
by other noncovalent bonding interactions, and the peptide lysine sidechains
are generally pointing away from the protein.
Contents
Close
Tutorials: How to See Cation-Pi Interactions in Protein Explorer
Contents
Close
- Tutorial: Interchain and intrachain amino acid pairs in 1b07.
- Click here to
start a new PE session with 1B07,
a small Src-Homology 2 (SH2) domain bound to a
regulatory peptide.
- Check "Cation-Pi" but not "Salt Bridges".
- Click the [Show] button.
After a short pause for processing, three cation-pi pairs will be
displayed. The default color scheme is to color by chain. This makes
it easy to see that two of the pairs are within the longer chain,
while one is between the peptide and the longer chain.
Note that you can distinguish Arg sidechains from Lys because the
cationic atoms of Arg are shown larger.
- Once you have displayed interacting pairs, you can change the
color scheme of these pairs immediately. Under Color
interacting pairs, change the radio buttons from "as below"
to "CPK" (color by element).
- Try another combination of the Display Options if you wish.
Click the [Show] button again.
(Click OK on the message that pops up.)
Advanced feature:
The message will suggest that you show the cation-pi pairs for
the originally selected 615 atoms -- the entire molecule.
When the cation-pi pairs are shown, Protein Explorer leaves
them selected. This makes it easy to change their rendering
or coloring using either QuickViews or typed commands.
However, if you re-run the cation-pi routine, you usually
want to show the pairs for the entire molecule.
- Comparison of the 3 pairs shown by Protein Explorer with the
results of
CaPTURE reveals that one of these three is deemed as
energetically insignificant.
- Reduce the maximum distance in Angstroms used in the
cation-pi routine from 6.0 to 5.5,
and click [Show] again. Only two pairs
meet the new distance cutoff, and these happen to be the two
deemed energetically significant by CaPTURE. However, it is
unusual for Protein Explorer's simple distance criterion to
agree so well with the results of CaPTURE.
- Increasing the cutoff distance to 6.5 Angstroms
reveals a 4th cation-pi pair (also deemed energetically
insignificant by CaPTURE).
- Way down at the bottom of the form, under the Advanced section,
check the Autostep feature.
In this mode, the detection distance will be incremented by 0.5 Angstroms
(or whatever you specify in the Autostep form slot) on each click of the
[Show] button.
Click the [Show] button several times, until you get to 8.5 Angstroms,
noting
the newly found moieties in each cycle.
This makes it easy to see that there are no more candidates for
energetically significant cation-pi pairs.
Contents
Close
- Tutorial: Cationic substrate interaction with enzyme in 2ACE.
- For this portion of the tutorial, you will need to load 2ace.pdb
(acetylcholinesterase with acetylcholine placed into the catalytic
gorge by modeling).
If you are connected to the Internet,
click here to
start a new PE session with 2ACE.
(If not, you will need to get a copy of 2ace.pdb on your hard disk
and load it with the Browse button on the Load Molecule page.)
- Check "Cation-Pi" but not "Salt Bridges", and make sure
"Autostep" (near the bottom of the page) is not checked.
- Click the link Restore Default Parameters in the
Cation-p section of the form, and accept the
confirmation by clicking OK.
- Click the [Show] button. Nine pairs
are shown. In this instance, Protein Explorer's distance-based
criteria do particularly badly: CaPTURE reports 12 energetically
significant cation-pi interactions; Protein Explorer misses
4 of these, and shows one deemed energetically insignificant
by CaPTURE. (By increasing the distance criterion to 6.5 Angstroms,
three of the missing pairs are shown, at the expense of
four new but energetically insignificant pairs also being shown.)
- Replace the contents of the cations slot with "ach.n1" (the
cationic nitrogen in the substrate).
Click the [Show] button.
Trp84 and Phe380 are shown as candidates for cation-pi interactions
with the substrate. Increasing the cutoff distance to 8.0 Angstroms
shows Phe331, but it is oriented with the edge of the ring
facing the cation.
Contents
Close
- Tutorial: Cationic ligand interaction with antibody in PROBLEMATIC PDB file 2mcp.
- 2mcp is the Fab fragment of an antibody bound to the hapten phosphocholine.
This case is problematic because of a bug in Chime 2 that prevents it
from correctly handling residues with names shorter than 3 characters
In this instance, the phosphocholine is named "PC" in the PDB file.
(The bug is that after selecting "PC", Chime fails to report its residue name
with the "show residue" command.)
Until a new version of Chime is released in which this bug is
fixed, the only solution to this problem is to save the PDB file
to your hard disk and edit it with a text editor, being careful
to save it as plain/ASCII/DOS text. You must give any residues of interest
three-character names. In this case, renaming PC to PC1 works fine.
Be careful to place the 3-character name in columns 18-20, so it lines
up with other 3-character residue names in the PDB file.
For your convenience, the PDB file so-modified has been made
placed on our server, and you can load it into Protein Explorer
by clicking here:
http://www.umass.edu/microbio/chime/beta/pdb/2mcp-pc1.pdb
- Check "Cation-Pi" but not "Salt Bridges", and make sure
"Autostep" (near the bottom of the page) is not checked.
- Click the link Restore Default Parameters in the
Cation-p section of the form, and accept the
confirmation by clicking OK.
- Change the contents of the cations slot to
"[pc1].n1". (Chime requires square brackets around residue names
that contain numerals.)
- Click the [Show] button.
Trp107 and Tyr100 are shown in favorable positions
for cation-pi interactions
with the hapten.
Contents
Close
- Tutorial: Aromatic ligand complex with enzyme in 3pcb.
- File 3pcb contains the enzyme dioxygenase complexed with 3-hydroxybenzoate.
Unfortunately it contains six copies of a heterodimer.
This produces images of unnecessary complexity.
For your convenience, a PDB file containing a single heterodimer (chains A, M)
has been made available, and can be loaded (if you are connected
to the Internet) by clicking here:
http://www.umass.edu/microbio/chime/beta/pdb/3pcb-am.pdb
- Check "Cation-Pi" but not "Salt Bridges", and make sure
"Autostep" (near the bottom of the page) is not checked.
- Click the link Restore Default Parameters in the
Cation-p section of the form, and accept the
confirmation by clicking OK.
- Protein Explorer needs to be told the names of three alternate carbons
in each type of aromatic ring for which it is to show proximal cations.
Clicking on the carbons in the 3HB ligand shows their names in the
message box.
Change the contents of the 3 slots for aromatic ring carbons to
these:
- [3hb].c1
- [3hb].c3
- [3hb].c5
(Chime requires square brackets around residue names
that contain numerals. You could equally well use carbons 2, 4, and 6.)
- Click the [Show] button.
Arg333 and Arg457 are shown in favorable positions
for cation-pi interactions
with the two copies of hydroxybenzoate.
Contents
Close
Protein Explorer's Distance-Based Search Mechanism for Cation-Pi Interactions
Chime has built-in at its disposal only distance, and the ability to
identify specific atoms within specific residues,
to use in searching for cation-pi interactions.
Gallivan and Dougherty considered potential interactions out to a distance
of 10 Angstroms, yet found that 99% of significant cation-pi interactions
occurred at distances not exceeding 6 Angstroms. In distances,
both NZ and CE of Lys, and both CZ and CD of Arg were considered.
The method built into Protein Explorer locates all cationic atoms
(by default, lys.nz, lys.ce, arg.cz, arg.cd) within (by default) 6.0 Angstroms
of three alternate carbons in a six-carbon ring of Trp, Tyr or Phe.
Advanced Explorer allows customization of the list of cationic atoms
considered, the list of aromatic atoms considered, or the distance.
This enables, for example, a bound substrate to be included in the
display. (The entire Chime script employed can be displayed.)
There are two reasons for overestimation by the present
distance-based method employed in Protein Explorer. In some
cases, a cation is within the requisite 6 Angstroms of an
aromatic sidechain, but the interaction would in fact be
energetically insignificant. In other cases, the requirement for
three alternate carbon atoms in aromatic rings are met by carbons
from different residues.
The latter type of
incorrect results are usually obvious because a ring will be
shown with no proximal cation, or vice versa.
(The effort that would be needed to
correct this flaw in the method seemed not worthwhile,
considering that interactions would still be overestimated due to
the lack of an energetics calculation).
Detailed comparisons of
results are given below.
Contents
Close
Comparison of Results from Protein Explorer vs. CaPTURE.
A goal for the future is to connect CaPTURE with Protein Explorer so that
CaPTURE's results can be visualized directly.
For the present, the simplistic distance-based method used in Protein Explorer
(when used with default parameters) in a few cases agrees
perfectly with the results of CaPTURE (1bl8, 2wea), but in most
cases shows more cation-pi pairs than the number of
energetically significant cation-pi interactions reported by
CaPTURE.
Occasionally the number of excess pairs can exceed the total
number of pairs reported by CaPTURE (5 added to 4 in 1bfg).
Detailed comparisons for a few cases are listed in a table below.
Occasionally, Protein Explorer fails to show an interaction reported
by CaPTURE (1 out of 10 in 1axi; 1 out of 14 in 1gai; 1 out of 6 in 2vab).
CaPTURE usually assigns barely significant
energies to the pairs not shown by Protein Explorer. They can be shown
by Protein Explorer if the default distance of 6.0 Angstroms is increased
in Advanced Explorer (at the expense of showing more energetically
insignificant pairs).
Reducing the distance (e.g. to 5.5 Angstroms) in some cases bring
Protein Explorer's results into agreement with those of CaPTURE (1v39),
but in others increases the number of energetically significant pairs
missed, while still finding some energetically insignificant pairs (1bfg).
Therefore, the default distance was left at 6.0 Angstroms.
This table displays best in a window at least 1024 pixels wide.
PDB ID
Resol. |
CaPTURE's Results* |
PE's Results* |
Total
Discrepancies** |
Comments |
1axi
2.1A |
AA 3 (1 1 0 0 1 0)
BB 6 (2 1 2 0 0 1)
AB 1 (0 0 1 0 0 0)
10 (3 2 3 0 1 1) |
AA 5 (2 2 0 0 1 0)
BB 11 (2 2 2 1 2 2)
AB 1 (0 0 0 0 0 0)
16 (4 4 2 1 3 2) |
AA 2 (-0, +2)
BB 5 (-0, +5)
AB 1 (-1, +0)
8 (-1, +7) |
The one miss is the least energetically significant pair (-2.72).
From the K179 end: CPCPCPc (only one excess hit, lower case).
|
1bfg
1.6A |
4 (2 1 0 0 1 0) |
9 (5 1 0 0 3 0) |
5 (-0, +5) |
Least energetically significant pair -2.16. |
1bl8
3.2A |
4 (0 0 4 0 0 0) |
4 (0 0 4 0 0 0) |
0 (-0, +0) |
Least energetically significant pair -7.80. |
1gai
1.7A |
14 (1 3 5 0 3 2) |
14 (1 2 6 0 3 2) |
2 (-1, +1) |
The one miss is the least energetically significant pair (-2.10) |
1v39
1.8A |
4 (2 2 0 0 0 0) |
7 (2 3 0 2 0 0) |
3 (-0, +3) |
Least energetically significant pair -4.34. |
2ace
2.5A |
12 (4 0 2 2 1 3) |
9 (4 0 0 3 1 1) |
5 (-4, +1) |
The one miss is the least energetically significant pair (-3.36) |
2vab
2.5A |
5 (0 2 3 1 0 0) |
8 (1 3 3 1 0 0) |
4 (-1, +3) |
The one miss is the least energetically significant pair (-3.36) |
2wea
1.25A |
0 (0 0 0 0 0 0) |
0 (0 0 0 0 0 0) |
0 (-0, +0) |
|
* Total pairs (counts of pairs RF RY RW KF KY KW).
** (-) missed pairs; (+) excess pairs.
Contents
Close
Inaccurate results: Rings Displayed without Cations and Vice Versa
If a ring is highlighted with no cation nearby, or vice versa, this
is consistent with the method employed. The explanation is as follows.
- In order for a ring to be shown, three of
its alternate carbons must be within 6.0 Angstroms of cation atoms. However,
the cation atoms need not be in the same residue. If no cation atom is
within 6.0 Angstroms of all three ring carbons, then no cation atom will
be shown. Example: in 4enl, Tyr6 is shown but no nearby cation atom
is shown. All three ring carbons in Tyr6 are within 6.0 Angstroms
of cations in Lys4 or Arg8; but none of the cation atoms is within 6.0
Angstroms of all three ring carbons. Having identified the lone ring
as Tyr6 (by clicking on it), the nearby cation atoms can be displayed
with commands such as
select within(6.0, tyr6) and (lys,arg)
bs
coe
("bs" is an alias for ball and stick; "coe" is an alias for color
by element (CPK).)
- In order for a cation atom to be shown, three alternate carbons
of a six-atom aromatic ring (in Trp, Tyr or Phe) must be within 6.0
Angstroms. However, the routine employed does not require that all three
atoms be in the same ring. Example: in 1gai, ARG273.CZ is shown
proximal to Trp228 and Phe267. It is not within 6.0 Angstroms
of all 3 alternate carbons in either ring, but it is within 6.0 Angstroms
of one atom in each group tested, which are by default:
- trp.cd2, tyr.cd1, or phe.cd1
- trp.cz2, tyr.cd2, or phe.cd2
- trp.cz3, tyr.cz, or phe.cz
So its three required
carbons come from the two different rings of Trp228 and Phe267.
Inaccurate results: Misoriented rings.
Another inappropriate result occurs in
1fod, where Arg114 is shown proximal to Phe39, but the
cation is in the plane of the ring instead of sitting on one
face. Phe39 is shown because it is close to another cation,
Arg179; it is not close enough to Arg114. Arg114 is shown because
three alternate ring carbons are close enough, but not in the
same residue. Some are in Phe39, and others are in Tyr11 (not
shown).
Contents
Close
Acknowledgements
Thanks to Joel Sussman (Weizmann Institute of Science,
Rehovot, Israel) for reminding me of the importance of cation-pi
interactions, providing me with the relevant literature, and
general support and encouragement.
Thanks also to Ricky Cox (Georgia Southern U, Statesboro GA)
for calling cation-pi interactions to my attention.
Contents
Close
Salt Bridge Detection
The default distance used for detection of salt bridges is 4.0 Angstroms
(suggested by Ricky Cox, Murray State U, KY).
This can be changed in the form slot.
At the bottom of the form is a checkbox for Autostep. This
causes the detection distance to be incremented by 0.5 Angstroms on each
click of the [Show] button. (The increment size can also be changed
in a form slot.) Ions newly detected in each cycle are highlighted with
a dot surface.
If you believe the default distance for salt bridge detection should be
different than 4.0 Angstroms, or if you know of published summaries of
the properties of protein salt bridges, please
enlighten me!
Contents
Close
Tutorial: Detecting Ligand Salt Bridges to Protein
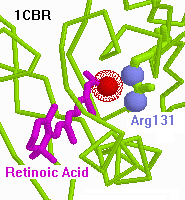
- You will need to load 1cbr.pdb
(cytoplasmic retinoic acid-binding protein complexed to retinoic acid).
If you are connected to the Internet,
click here to
start a new PE session with 1CBR.
(If not, you will need to get a copy of 1cbr.pdb on your hard disk
and load it with the Browse button on the Load Molecule page.)
- Check "Salt Bridges" but not "Cation-Pi", and make sure
"Autostep" (near the bottom of the page) is not checked.
- Click the link Restore Default Parameters in the
Cation-p section of the form, and accept the
confirmation by clicking OK.
- Click on the carboxyl oxygens in the retinoic acid to show
their atom and residue names in the message box.
- In the Salt Bridges form, replace the anion atoms in the
form slot with "rea.o?" (letter o for oxygen, not numeral; the
question mark matches either "rea.o1" or "rea.o2").
- Click the [Show] button. Arg 131 is identified
as within salt bridge distance of the ligand anion.
- Feel free to try the "Autostep" mode to look for more distant
cations, or restore the default anions and look for within-protein
salt bridges.
Contents
Close